
 会员中心
会员中心 EN
EN

 2025.12.11.
2025.12.11.
低磷性佝偻病
什么是低磷性佝偻病?
低磷性佝偻病(Hypophosphatemic Rickets,简称HR)是一种较为罕见的疾病,发病率约为1/25,000。它主要是由于遗传或后天因素导致肾脏排磷过多,进而引发血磷水平严重低下以及骨骼矿化障碍。儿童期表现为方颅、鸡胸、肋骨串珠、四肢弯曲畸形(O型腿或X型腿)、生长迟缓、身材矮小、步态摇摆、牙齿发育异常(牙釉质破坏、反复牙脓肿)等;成人期则表现为骨痛、肌无力、多发性骨折、身材变矮、活动受限等。
发病机制
低磷性佝偻病主要分为遗传性和获得性两大类:
一)遗传性类型
X连锁显性遗传低磷性佝偻病(XLH):由PHEX基因突变导致的,最常见的遗传类型,约占遗传性低磷性佝偻病的80%。
常染色体显性遗传低磷性佝偻病(ADHR):由FGF23基因突变引起,最常见的突变位点为 c.527G>A (p.Arg176Gln)。
常染色体隐性遗传低磷性佝偻病(ARHR):包括DMP1、ENPP1、FAM20C等基因突变亚型。
遗传性低磷血症伴高钙尿症(HHRH):由SLC34A3基因突变导致。
二)获得性类型
肿瘤性骨软化症(TIO):由某些良性肿瘤分泌过多FGF23引起。
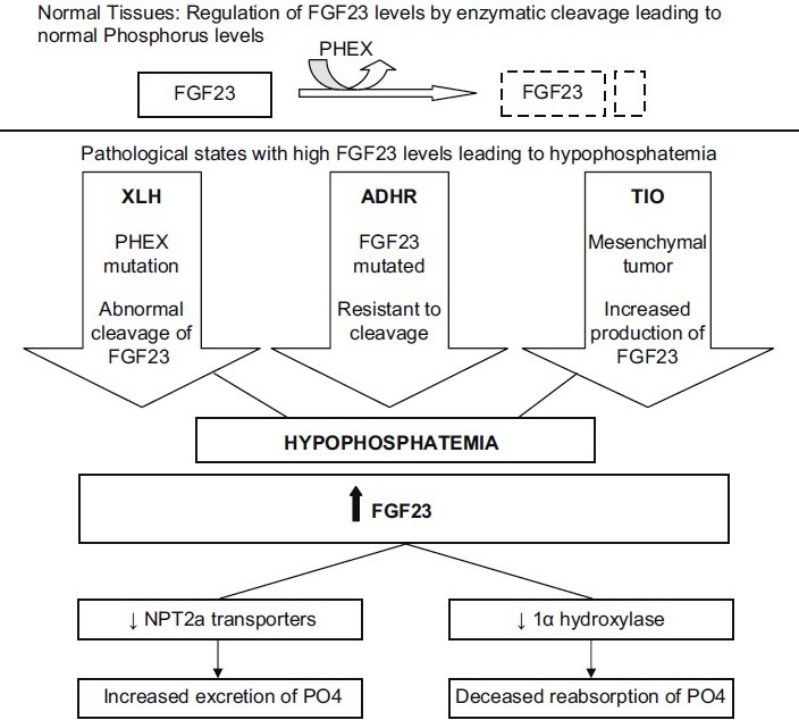
图源:Hypophosphatemic rickets
在多数类型中,低磷性佝偻病的核心发病机制涉及FGF23-PHEX轴的异常调节。成纤维细胞生长因子23(FGF23)是磷代谢的核心调节因子,由骨细胞和成骨细胞分泌。PHEX基因位于X染色体p22.1上,其编码的PHEX蛋白主要在骨细胞和成骨细胞表达。
在正常生理状态下,PHEX蛋白通过与DMP1等N-连接糖蛋白(SIBLING蛋白)相互作用,稳定一个抑制FGF23转录的复合物,并防止具有抑制矿化作用的ASARM肽的释放,从而抑制FGF23的表达。然而,当PHEX基因发生突变而失活时,一方面导致ASARM肽积累,直接抑制骨矿化并上调FGF23表达;另一方面使骨细胞中FGF23的转录和合成显著增加。升高的FGF23会负向调节血清1,25-二羟维生素D[1,25-(OH)?D]水平,进而抑制肾近端小管和肠道对磷的重吸收,同时减少肠道磷的吸收,最终导致持续的低磷血症,引发佝偻病或骨软化症的骨骼临床表现。
基因治疗
小环DNA(MC-DNA)治疗:2025年10月,山东省立医院徐潮、赵家军团队在《Advanced Science》发表的研究显示,使用表达FGF23片段(氨基酸180-251)的小环DNA载体治疗XLH小鼠模型,可显著改善血磷水平、降低血清碱性磷酸酶,并改善骨矿化,且至少6周内未观察到明显不良反应。
腺相关病毒(AAV)载体治疗:表达FGF23 C端尾部的肝靶向AAV载体可改善XLH小鼠模型的骨骼表现和骨软化症。
小鼠模型
DMP1-KO小鼠:敲除DMP1基因,模拟常染色体隐性遗传低磷性佝偻病(ARHR1),该模型表现为FGF23水平显著升高、严重低磷血症及骨矿化缺陷。
SLC34A3-KO小鼠:敲除Slc34a1基因,模拟遗传性低磷血症伴高钙尿症(HHRH),表现出血磷水平异常和骨骼病变。
Hyp小鼠:这是一种经典的低磷性佝偻病模型,其体内PHEX基因发生突变,导致FGF23表达异常,从而引发一系列与人类低磷性佝偻病相似的症状。
PHEX-T1349C敲入小鼠:通过在PHEX基因中引入T1349C突变,能模拟特定PHEX基因突变导致的低磷性佝偻病。
FGF23-R176Q小鼠:FGF23基因发生R176Q突变,导致FGF23功能异常,进而引起磷代谢紊乱和骨骼病变。
明迅生物助力基因治疗
基因治疗为罕见病带来了希望,但其研发和验证离不开动物模型的支持。明迅生物借助自主研发的TurboMice™技术已研发了多个罕见病小鼠模型,TurboMice™技术突破了小鼠造模周期长和复杂模型成功率低的技术难题,可实现几乎任何目标基因位点的编辑,可短至2个月由胚胎干细胞直接制备完整纯合基因编辑小鼠模型。
明迅生物可根据客户需求定制各类HR小鼠模型,DMP1-KO小鼠、SLC34A3-KO小鼠、Hyp小鼠、PHEX-T1349C敲入小鼠、FGF23-R176Q小鼠等,欢迎各位老师前来咨询。
参考资料:
[1]丁桂霞. 低磷性佝偻病研究新进展[J]. 中华实用儿科临床杂志,2019,34(17):1304-1308.DOI:10.3760/cma.j.issn.2095-428X.2019.17.006
[2]Jagtap VS, Sarathi V, Lila AR, Bandgar T, Menon P, Shah NS. Hypophosphatemic rickets. Indian J Endocrinol Metab. 2012 Mar;16(2):177-82. doi: 10.4103/2230-8210.93733. PMID: 22470852; PMCID: PMC3313733.
[3]Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006 Feb;78(2):193-201. doi: 10.1086/499410. Epub 2005 Dec 9. PMID: 16358215; PMCID: PMC1380229.
[4]Jagtap VS, Sarathi V, Lila AR, Bandgar T, Menon P, Shah NS. Hypophosphatemic rickets. Indian J Endocrinol Metab. 2012 Mar;16(2):177-82. doi: 10.4103/2230-8210.93733. PMID: 22470852; PMCID: PMC3313733.
特别声明:本文来自明迅生物公众号,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。转载授权请在公众号后台留言联系我们。其他合作需求,请联系sales@mingceler.com。
免责声明:部分素材源于网络,如有侵权,联系删除,本文仅作信息交流之目的,亦不是提供治疗方案,文中观点不代表明迅生物立场,亦不代表明迅生物支持或反对文中观点。

