
 会员中心
会员中心 EN
EN

 2025.11.06.
2025.11.06.
什么是Rett综合征?
Rett综合征(Rett syndrome,RTT)是一种由X染色体上的甲基化CpG结合蛋白2(MECP2)基因突变引起的严重神经系统疾病,主要影响女性,通常在出生后6到18个月之间开始显现。患者在出生后初期有短暂的正常发育,随后出现功能倒退,包括语言和手部功能的丧失、手部刻板动作、步态异常、呼吸节律不整(呼吸暂停、过度换气)、脊柱侧弯、焦虑和睡眠障碍等,RTT发病几率约为1/10,000 至 1/15,000。
发病机制
Rett综合征的核心发病机制与MECP2基因功能丧失性突变密切相关。该基因编码的甲基-CpG结合蛋白2(MeCP2)是一种至关重要的表观遗传调控因子,通过识别DNA甲基化标记,动态调控数千个下游基因的表达。一方面,MeCP2通过招募组蛋白去乙酰化酶等共抑制复合物,使染色质结构紧缩,抑制基因转录;另一方面,它也能通过与转录激活因子相互作用直接促进基因表达,如对脑源性神经营养因子(BDNF)的调控。在神经发育过程中,MeCP2维持精确的基因表达程序,对神经元的成熟、树突棘形态、突触可塑性及神经环路的兴奋/抑制平衡起核心的“总开关”作用。
MECP2基因突变导致MeCP2蛋白功能受损,其转录调控功能的丧失造成全基因组表达失调,进而损害突触结构与功能,特别影响GABA能抑制性神经元,最终导致大脑关键神经网络功能异常,出现发育倒退、运动障碍、癫痫和自主呼吸失调等典型症状。在Rett综合征中,最常见的MECP2基因突变主要集中在几个特定的“热点”位点,如p.Arg168X、p.Arg255X、p.Arg270X、p.Arg294X等无义突变,以及p.Thr158Met (T158M)、p.Arg106Trp (R106W)、p.Arg133Cys (R133C)和p.Arg306Cys (R306C)等错义突变。
除MECP2外,CDKL5和FOXG1等基因的突变也可引起非典型Rett综合征。CDKL5作为一种激酶,可通过磷酸化修饰MeCP2来调节其活性,从而间接影响神经元功能;而转录因子FOXG1则在调控前脑发育中与MeCP2存在通路交叉。这些基因的突变通过干扰共同的下游网络,导致了与RTT相似的神经发育障碍表型。
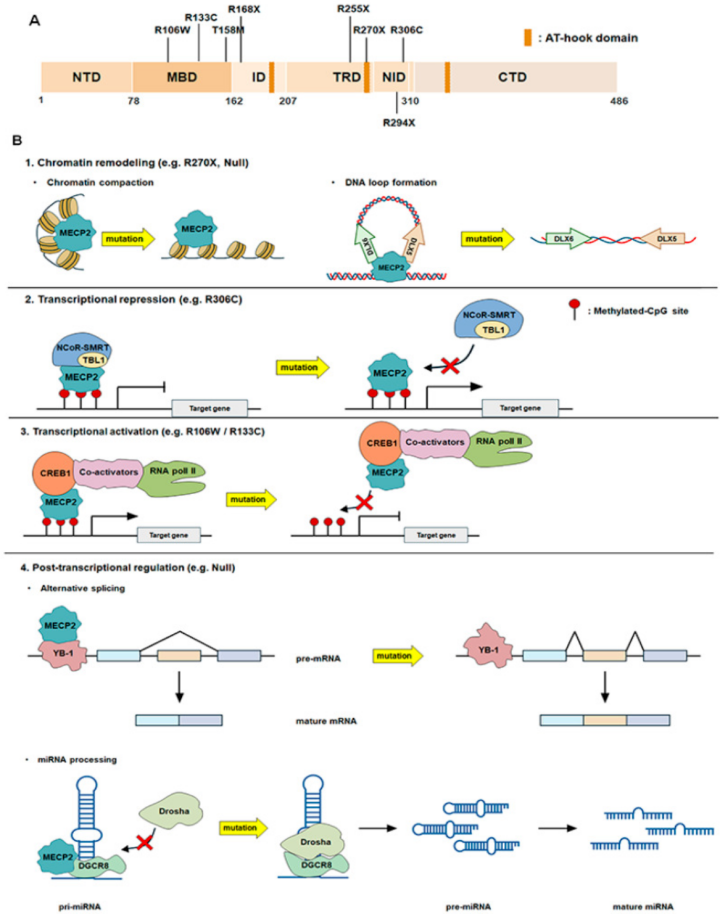
图源MECP2 Dysfunction in Rett Syndrome: Molecular Mechanisms, Multisystem Pathology, and Emerging Therapeutic Strategies
基因治疗
•AAV基因疗法:2025年3月,广州妇儿中心完成了中南地区首例Rett综合征基因治疗。该治疗通过AAV载体将正常的MECP2基因直接递送到患儿的大脑受损神经元中,修复神经细胞的功能,从而缓解患儿的神经发育障碍症状。
•A-to-I RNA编辑:通过A-to-I RNA编辑技术,将R270X无义终止突变转化为色氨酸,恢复MECP2功能。实验结果表明,这种编辑技术在小鼠模型中表现出良好的效果,且没有显著的行为差异。
•替代基因疗法:通过过表达一种转基因神经营养因子(TF)来改善RTT症状。研究者将TF的编码序列包装到AAV中,利用AAV-PHP.eB因其能有效穿过啮齿动物血脑屏障(BBB),将TF克隆到一个星形胶质细胞特异性启动子下游。实验结果显示,低剂量的AAV-TF治疗可以显著改善MECP2基因敲除小鼠的运动行为。
小鼠模型
MECP2 KO小鼠:完全敲除MECP2基因,模拟Rett综合征的病理特征。这些小鼠表现出类似Rett综合征的神经症状,如运动障碍、呼吸异常和早死。
MECP2 T158A小鼠:p.Thr158Ala突变,特异性破坏MeCP2蛋白的DNA结合能力,用于研究此功能缺失的影响。
MECP2 R306C小鼠:p.Arg306Cys突变,主要影响MeCP2蛋白招募转录抑制复合物的功能,用于揭示转录调控异常的作用。
Viaat-Mecp2小鼠:通过特异性敲除GABA能神经元中的MECP2基因,研究GABA能神经元在Rett综合征中的作用。
明迅生物助力基因治疗
基因治疗为罕见病带来了希望,但其研发和验证离不开动物模型的支持。明迅生物借助自主研发的TurboMice™技术已研发了多个罕见病小鼠模型,TurboMice™技术突破了小鼠造模周期长和复杂模型成功率低的技术难题,可实现几乎任何目标基因位点的编辑,可短至2个月由胚胎干细胞直接制备完整纯合基因编辑小鼠模型。
明迅生物可根据客户需求定制各类RTT小鼠模型,如MECP2 KO小鼠、MECP2 T158A小鼠、MECP2 R306C小鼠、Viaat-Mecp2小鼠等,欢迎各位老师来咨询!
参考资料:
[1]https://www.merckmanuals.com/professional/pediatrics/congenital-neurologic-anomalies/rett-syndrome
[2]Kyle SM, Vashi N, Justice MJ. Rett syndrome: a neurological disorder with metabolic components. Open Biol. 2018 Feb;8(2):170216. doi: 10.1098/rsob.170216. PMID: 29445033; PMCID: PMC5830535.
[3]吴浩, 钟敏. Rett综合征的分子遗传学和治疗研究进展[J]. 现代医药卫生, 2024, (11): [1945-1949]. DOI: 10.3969/j.issn.1009-5519.2024.11.030.
[4]Liyanage VR, Rastegar M. Rett syndrome and MeCP2. Neuromolecular Med. 2014 Jun;16(2):231-64. doi: 10.1007/s12017-014-8295-9. Epub 2014 Mar 11. PMID: 24615633; PMCID: PMC5798978.
[5]Lu S, Chen Y, Wang Z. Advances in the pathogenesis of Rett syndrome using cell models. Animal Model Exp Med. 2022 Dec;5(6):532-541. doi: 10.1002/ame2.12236. Epub 2022 Jul 4. PMID: 35785421; PMCID: PMC9773312.
[6]Choi G, Lee S, Yoo S, Do JT. MECP2 Dysfunction in Rett Syndrome: Molecular Mechanisms, Multisystem Pathology, and Emerging Therapeutic Strategies. Int J Mol Sci. 2025 Aug 26;26(17):8277. doi: 10.3390/ijms26178277. PMID: 40943197; PMCID: PMC12428351.
特别声明:本文来自明迅生物公众号,欢迎个人转发至朋友圈,谢绝媒体或机构未经授权以任何形式转载至其他平台。转载授权请在公众号后台留言联系我们。其他合作需求,请联系sales@mingceler.com。
免责声明:部分素材源于网络,如有侵权,联系删除,本文仅作信息交流之目的,亦不是提供治疗方案,文中观点不代表明迅生物立场,亦不代表明迅生物支持或反对文中观点。
明迅生物:模式小鼠黑科技

